
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 |
- Python
- 선형대수
- 오블완
- scanpy
- 대학생재테크
- endothelial
- single cell rna sequencing
- 대학원
- 대학생주식
- journal meeting
- 넘파이 매서드
- singe cell ran sequencing
- 후기
- liver
- np.flatten
- R
- np.diagflat
- pcrnaseq
- np.triu
- 주식
- bioinformatics
- single cell rna sequening
- scRNASeq
- 통계학
- Tutorial
- 주식투자
- np.trace
- numpy
- Preprocessing
- 티스토리챌린지
- Today
- Total
biotechknowledge
[데이콘]2024 생명연구자원 AI활용 경진대회 : 인공지능 활용 부문 상위 5% (후기, 데이터전처리 방식, trouble shooting) 본문
[데이콘]2024 생명연구자원 AI활용 경진대회 : 인공지능 활용 부문 상위 5% (후기, 데이터전처리 방식, trouble shooting)
준2준2 2024. 11. 24. 13:36
SKN AI 캠프에서 만난 친구들과 Dacon 대회에 참가하였다. 우연히도 바이오데이터를 활용한 인공지능 대회가 있었고 배경지식을 이용해서 AI 적용해볼 기회를 가질 수 있었다. 나는 주로 데이터 전처리를 담당했고 팀원들이 다양한 머신러닝 기법을 적용해보면서 성능을 끌어올리는데 집중했다. 간단하게 데이터와 어떤 방식으로 접근하고 발전시켰는지 정리해 보겠다.

먼저 주어진 데이터이다. 데이터를 보면 아미노산 변이정보가 HGVS (Human Genome Variation Society) 표기법으로 나와있다. 정상인은 WT(wild type) 변이가 생긴것은 어떤 아미노산이 어디 위치에서 어떤 아미노산을 바꼈는지 나타낸다. 예를들어 R895R은 아미노산 R이 895위치에서 아미노산R로 변이된 것. 즉 침묵돌연변이로 염기서열하나가 바뀌긴 했지만 단백질에는 변화가 없음을 알 수 있다. (돌연변이 종류, 아미노산 종류 까지 다 설명하면 너무 길어지니까 생략)
유전자의 종류는 열에, 환자하나한의 샘플은 행에 나와있다. (이 데이터가 실제 암환자 데이터라고 생각한다. 가짜로 만들어낸 데이터라면 현업에 적용할 수 도 없을 뿐더러 돈까지 걸어가며 대회를 하는 이유가 없기 떄문이다.) SUBCLASS 열을 보면 암종을 볼 수 있다.
import matplotlib.pyplot as plt
# 암종별 개수를 계산
subclass_counts = train['SUBCLASS'].value_counts()
# 히스토그램 생성
plt.figure(figsize=(12, 6))
plt.bar(subclass_counts.index, subclass_counts.values, edgecolor='black', color='orange')
plt.xticks(rotation=90) # x축 레이블 회전
plt.title('Cancer Subclass Distribution', fontsize=14)
plt.xlabel('Subclass', fontsize=12)
plt.ylabel('Count', fontsize=12)
plt.tight_layout() # 레이아웃 조정
plt.show()
클래스불균형이 심한 dataset, 실제 암종별 발병률이 다르니 그럴 수 있다.
접근1. Qualitative Data -> numeric value
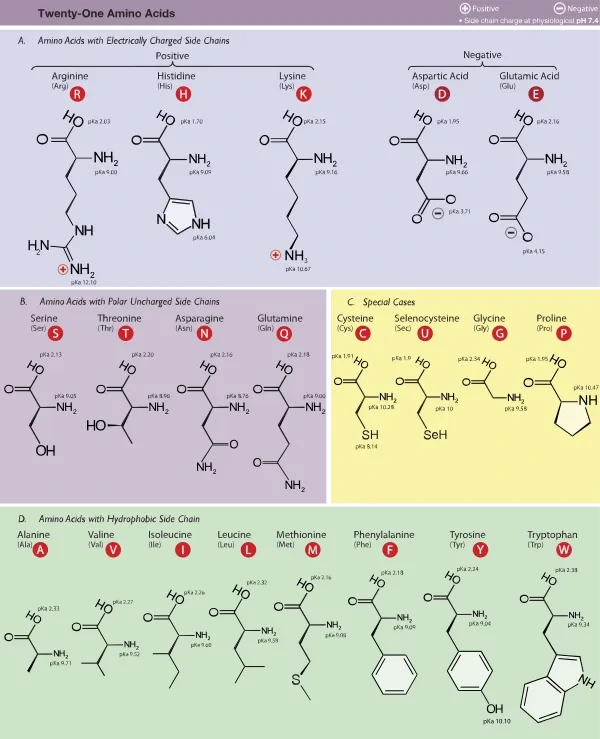
머신러닝을 돌리기위해선 아미노산 변이정보를 양적자료로 바꿔줘야 한다. 아미노산 변이 치명도에 따라서 심할수록 높은 점수를 주고 침묵돌연변이와 같은 단백질의 기능에 문제가 없는 경우는 낮은점수를 부여하는 것이다. 아미노산은 생화학적으로 그룹화할 수 있다. 교과서별로 혹은 그룹짓는 기준에 따라서 디테일한 분류가 달라지긴 하지만 큰 틀에서는 변화가 없다. 예를들어 위에 표에서 D panel(초록색)에 속하는 아미노산 그룹내에 속한 변이는 타 그룹으로 변이되는 것보다 치명도가 낮다고 판단하는 것이다.
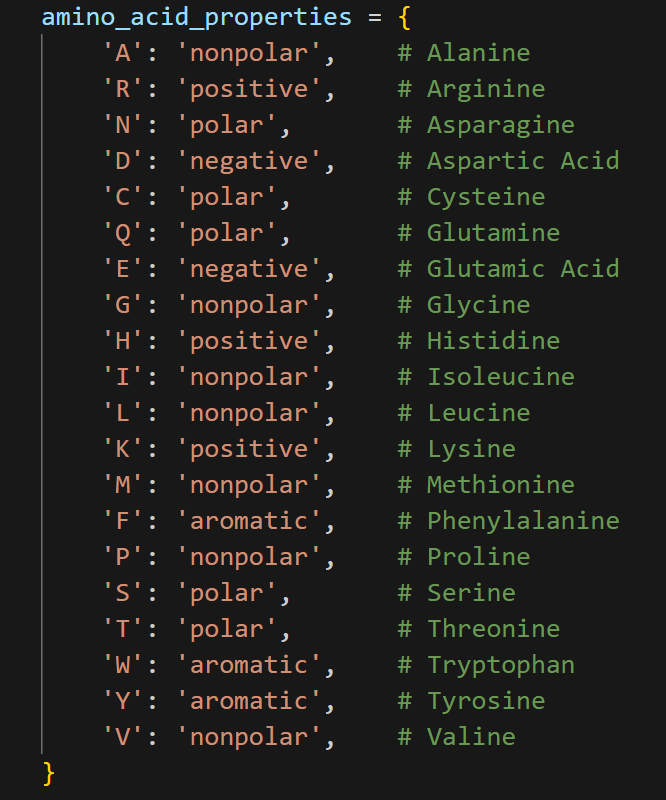
GPT를 이용해서 아미노산을 그룹으로 분류하엿다. nonpolar, polor , negative, positive, aromatic 으로 구분하여 같은 그룹끼리의 변이는 낮은 점수, 타 그룹으로 변이는 높은 점수를 부여 하였다.
접근2. 외부데이터 활용
접근 1로 한차례 성능을 끌어올리는데 성공했다. 치명도점수를 이리저리 조절해보며 최적의 가중치를 찾으려 노력했다. 예를들어, 아미노산 변이 치명도의 맨 위에는 frame shift, stop, insertion 등 이 있다. 이런 변이들을 30으로도 해보고 60으로도 해보고 침묵돌연변이, 비동의 돌연변이, 각각의 치명도 점수와의 관계도 고려해서 튜닝을 계속해봤지만 크게 달라지지 않았다.
데이터셋과 인공지능의 목적 자체는 생소한 주제가 아니다. 누구나 다 생각해보고 이미 수백,수천개의 그룹에서 시도해봤을거라 생각하고 관련논문을 찾아보았다. 그러다 우연히 driver gene, slient gene 을 분류해놓은 데이터베이스를 발견하였다. 어떤 유전자에서 어떤 변이가 암을 발생시키더라, 암을 발생 안시키더라 하는 정보이다.
놀라웠던점은 공동의 목표(인간의 암 정복)을 위해 수십년간 쌓인 데이터베이스가 공유되고 있었다. 그것도 데이터 종류도 다양하고 여러 사이트가 많았는데 이번 대회에서는 COSMIC 데이터를 간접적으로나마 사용해 볼 수 있었다. 데이터는 충분하다. 내 실력이 부족해서 그렇지.
https://www.sciencedirect.com/science/article/pii/S0925443915003361
Exploring preferred amino acid mutations in cancer genes: Applications to identify potential drug targets
Somatic mutations developed with missense, silent, insertions and deletions have varying effects on the resulting protein and are one of the important…
www.sciencedirect.com
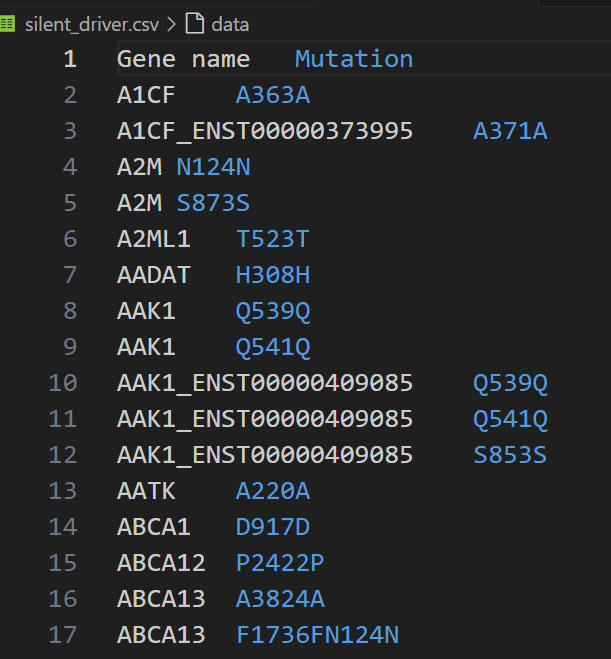
위는 silent_driver.csv 의 일부이다. A>A로 아미노산 변이가 일어나지 않아 치명도가 낮게 부여되고 있었다. 하지만 위의 silent_driver의 의미는 아미노산이 변이가 없음에도 암발생을 일으킨다는 의미이다. 3% 내외의 성능향상이 있었지만, 외부데이터의 크기가 크지 않아서 드라마틱한 성능향상은 아니었다.
접근 3. blosum62
성능향상에 한계가 느껴지자, 아미노산 변이 치명도를 더 세분하게 나눠보고자 했다. 아미노산의 종류는 20개이다. 변이가 가능한 총 경우의 수는 20X20 = 400 개이다. 이 400개의 변이 각각의 치명도를 고려해서 점수를 부여하는 방법을 찾아보았다.
http://www.insilicase.com/Web/SubstitutionScore.aspx
Mutation severity
Insilicase deus ex computa
www.insilicase.com
그러던 중 위에 블로그(?) 같은 아마추어 연구자가 사용한 방식에 착안해서 blosum62 table 기반으로 아미노산 치명도를 점수화해 보았다.
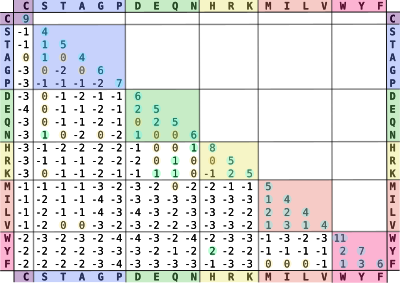
blosum62 는 진화적으로 축적된 데이터를 기반으로 어떠한 아미노산이 오래 유지되고 있는지 확인할 수 있다. 숫자가 높을수록 단백질 기능에 핵심적이라고 생각할 수 있다. 예를들어 W(트립토판)은 아미노산 중에서도 독특한 모양을 하고 있다. 고리가 2개있고 크기도 굉장히 크다. 다른 아미노산으로 대체되기 힘들다. 이러한 특수성 때문에 트립토판의 변이는 생물에게 치명적일 수 있고 트립토판변이가 생긴 생물은 도태되기 쉽다. 즉, 트립토판은 오랜기간 생물의 특정 기능내에서 유지되고 있고 blosum62에서 가장 높은 점수인 11점을 갖는 것이다. 이 내용을 적용해서 테이블내 숫자가 높은 아미노산의 변이에 더 높은 가중치를 부여하였다. 하지만 성능향상은 크게 없었다.
blo_score = blo_score.applymap(lambda x: int(x))
for i in blo_score.columns:
blo_score[i] = blo_score.loc[i,i] - blo_score[i]
blo_score = blo_score.T
blo_score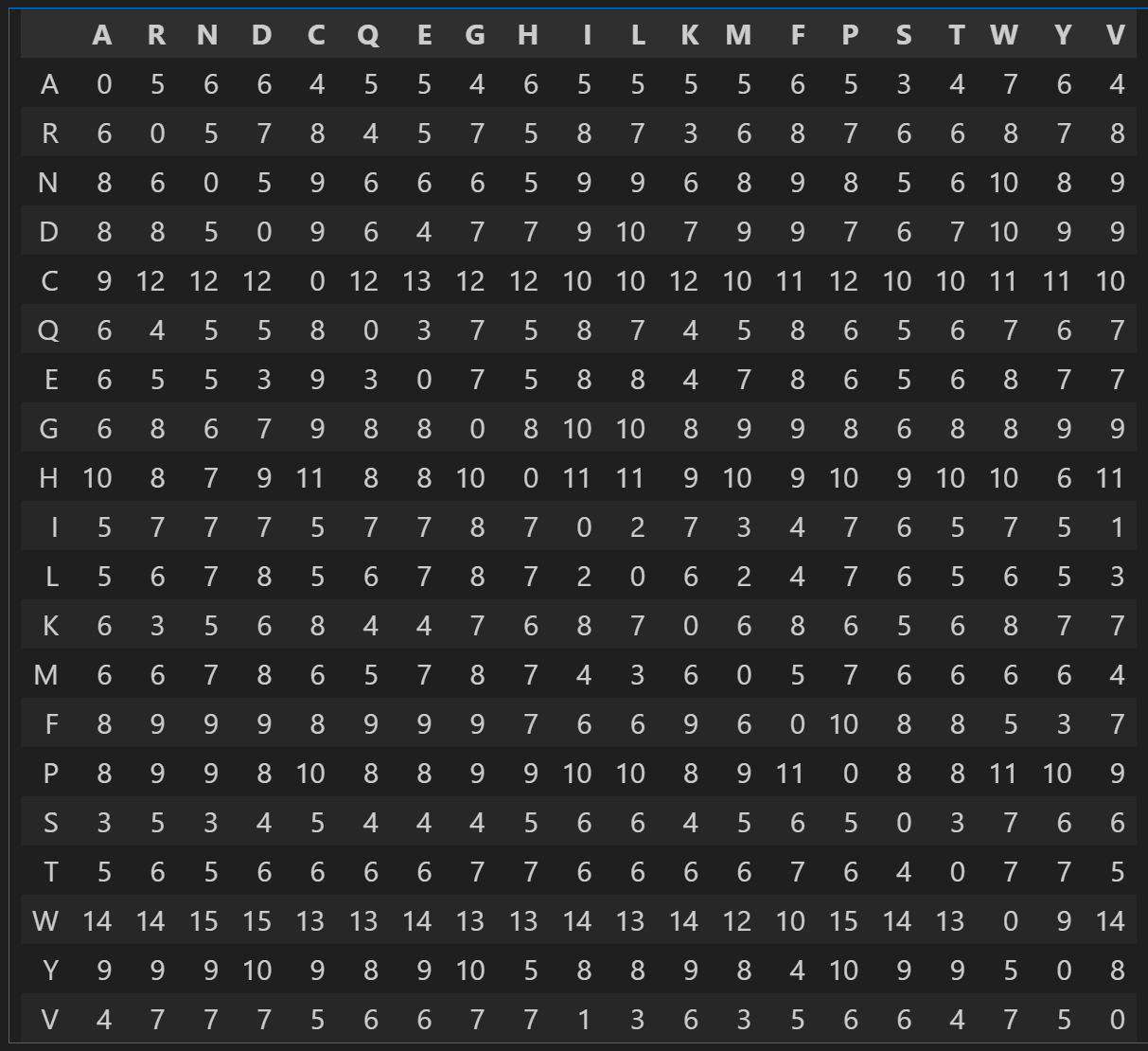
접근 4. 변이 위치정보 활용
어떻게 하면 성능을 더 끌어올릴지 고민하다가 위치정보를 이용하지 않고 있다는 사실에 주목했다. 변이정보는 나눌만큼 나누고 튜닝도 여러번 해봐서 더이상의 성능향상을 기대하기 어려웠다. 우리팀은 위치정보를 활용할 방법을 고민해보았다. 돌연변이 발생-위치는 종속적인 관계이기 때문에 무작정 돌연변이 발생위치만 따로 떼내어 컬럼을 만들 수는 없었다. 돌연변이 치명도 점수와 발생위치를 묶어내야 했고 고민하다가 이미지처럼 처러하면 할 수 있겠다는 생각이 떠올랐다.
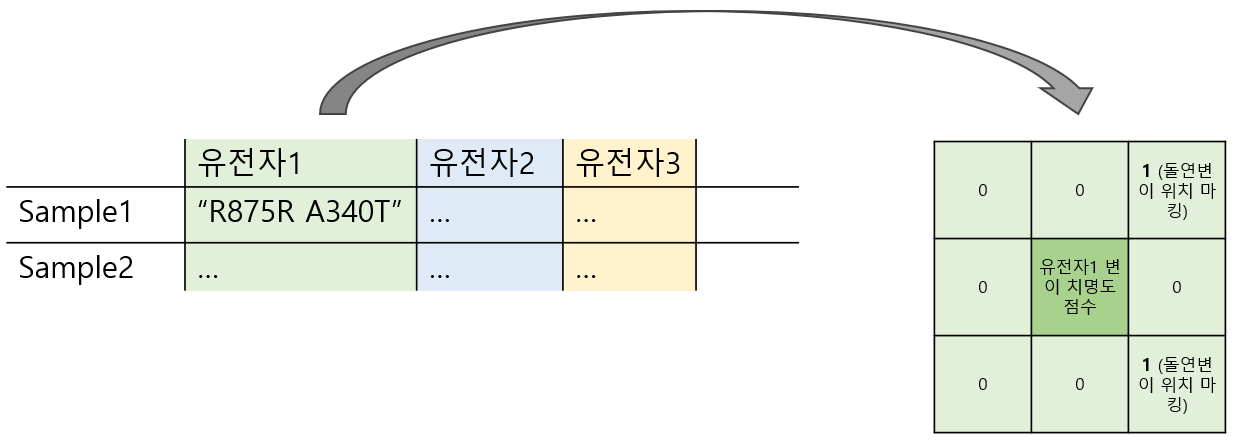
유전자 중 일부를 제거하고 총 4,225개의 유전자를 남겼다. ( train, test 에서 변이가 생기지않은 유전자들 제거) 마침 4,225개는 65X65 로 정사각행렬 모양으로 처리가 가능했다. 유전자1에 생긴 치명도 점수의 합을 가운데에 넣는다. 유전자 sequence를 8등분하여 치명도 점수를 둘러쌓는 배치를 만든다. 돌연변이 발생위치에 count up 한다. 예를들어, 유전자1에 에 2번의 돌연변이가 생겼을때 3번구간 8번구간에 변이가 생긴경우 위의 경우와 같이 3,8번 박스에 count 를 올린다.
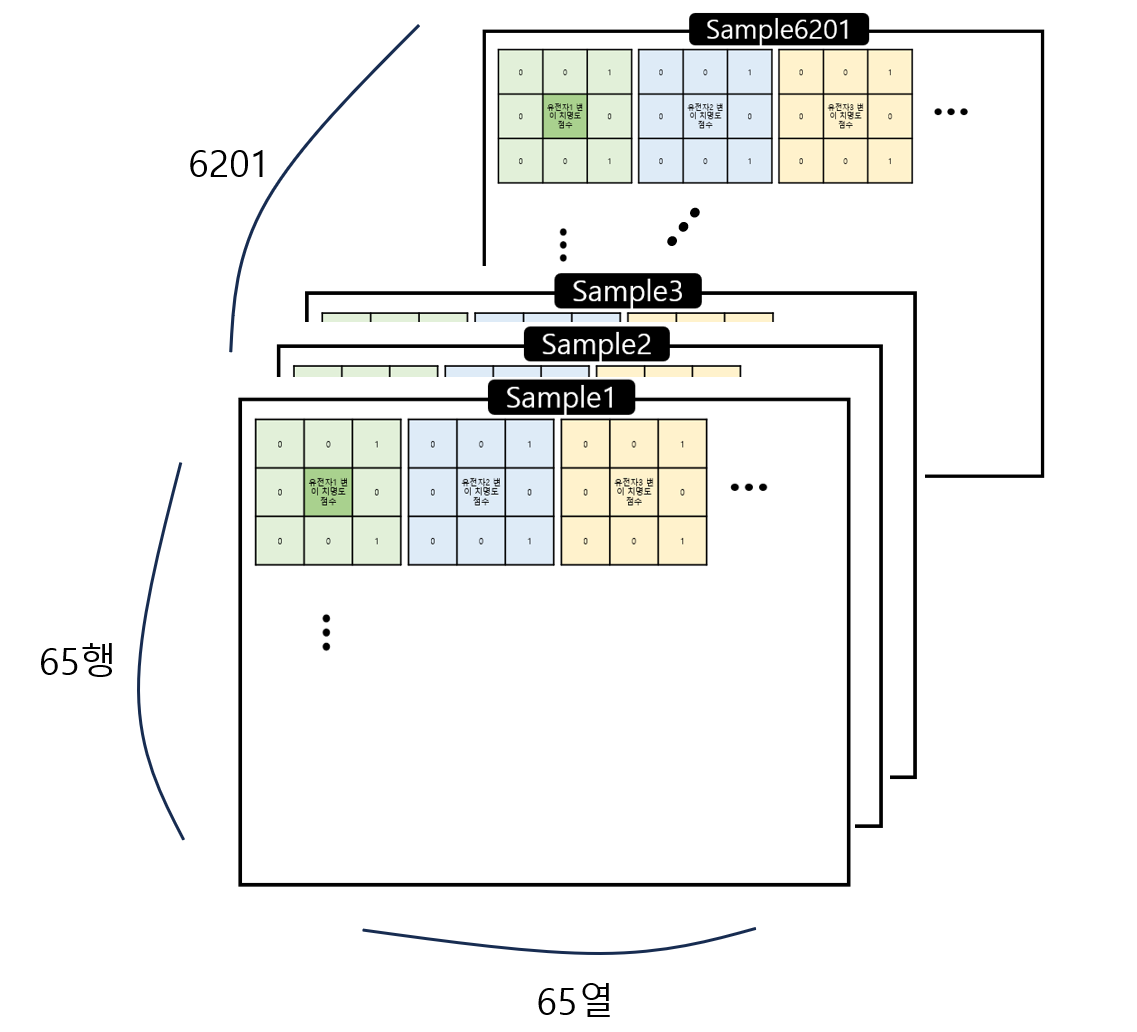
즉, 환자(Sample) 1개당 한장의 이미지가 만들어지고 총 6,201개의 이미지가 만들어지고 각각의 이미지는 65*65개의 유전자를 담고 있다. 이미지의 크기는 195 * 195 이다. (유전자 1개당 3 *3) 이 이미지로 CNN을 적용시킬 때 제일 첫 번째 층에서 Convolution을 할 때 slide 2를 주어 3*3 정사각행렬을 각각 독립적으로 학습시키는게 핵심이다.
import matplotlib.pyplot as plt
import numpy as np
# BRCA 클래스의 모든 이미지 가져오기
images = subclass_img_dict['BRCA']
# 모든 이미지를 평균화 (축 0 기준)
average_image = np.mean(images, axis=0)
# 평균 이미지를 시각화
plt.figure(figsize=(6, 6))
plt.imshow(average_image, cmap='viridis') # 노란색 계열
plt.colorbar() # 색상바 추가
plt.title('Average Image of BRCA Class', fontsize=14)
plt.axis('off') # 축 숨김
plt.show()
제일 smaple이 많았던 BRCA 의 평균이미지 이다. 인간이 보기에는 그냥 보라색 색종이지만 기계는 무언가 찾아낼 수 있지 않을까 기대했다. 아쉽지만 큰 성능에는 큰 차이가 없었다.
내가 시도해보았던 전처리는 여기까지이고 팀원들이 머신러닝 튜닝을 통해서 성능을 개선시켰다. 기반이 다른 알고리즘을 섞어서 스태킹한 모델이 성능이 좋았고, 스태킹과정을 AutoML로 최고성능 모델을 만들어서 최종 제출하였다.

개발 공부를 시작하면, 유튜브나 주변 경험자의 조언을 많이 찾아보곤 한다. 흔한 질문들을 보면 다 똑같은 맥락이다. '어떻게 공부해요?', '어떻게 하면 빨리 늘어요?', '뭐부터 시작해요?' 사실 그에대한 대답도 사실 비슷하다. 직접 해보는 것. 아주 작은 것부터 만들어보고 실행시켜보면서 문제해결력을 기르고 거기서오는 재미를 느끼는 것. 개발공부 2달 정도 했는데 위에 데이콘 참가하면서 보낸 3주동안 제일 많이 배웠고 재밌게 공부했다. 훌륭한 팀원들을 만나 많이 배울 수 있어서 운이 좋았다. they are all A class palyers.

스티브 잡스가 말하는 CLASS A PLAYER.