
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 |
- endothelial
- np.diagflat
- 통계학
- single cell rna sequening
- 오블완
- Preprocessing
- 주식
- 선형대수
- 대학생재테크
- 후기
- np.flatten
- bioinformatics
- liver
- journal meeting
- 주식투자
- single cell rna sequencing
- scanpy
- 넘파이 매서드
- scRNASeq
- np.triu
- R
- 대학원
- numpy
- 티스토리챌린지
- np.trace
- Tutorial
- pcrnaseq
- singe cell ran sequencing
- Python
- 대학생주식
- Today
- Total
biotechknowledge
Scanpy tutorials - preprocessing and clustering (anndata, pooch 개념설명) 본문
Scanpy tutorials - preprocessing and clustering (anndata, pooch 개념설명)
준2준2 2024. 11. 7. 18:34
# Core scverse libraries
import scanpy as sc
import anndata as ad
# Data retrieval
import pooch
AnnData single cell rna 데이터를 체계적으로 저장하기 위한 구조로, 유전자 발현 행렬을 X라는 속성에 저장하고, 유전자 ID와 세포 바코드 정보를 var(변수)와 obs(관측값)에 각각 저장한다.
obs (Observations):
- obs는 세포에 대한 메타데이터를 (실험 조건, 세포의 특성, 클러스터링 결과 등) 저장하는 곳입니다. 데이터프레임 형태를 가지며, 각 행이 하나의 세포를 나타냅니다.
- 예를 들어, 각 세포에 대해 클러스터 레이블, 샘플 조건, 배치 정보 등 다양한 정보를 이곳에 저장할 수 있습니다.
var (Variables):
- var는 유전자에 대한 메타데이터를 (유전자 이름, 기능적 주석, 유전자 발현 패턴, 유전자 기능 분류, 발현 위치 등) 저장하는 부분입니다. 데이터프레임 형식이며, 각 행이 하나의 유전자에 해당한다.
pooch - 데이터 파일을 지정된 URL에서 다운로드하고, 로컬 캐시에 저장합니다.
sc.settings.set_figure_params(dpi=50, facecolor="white")
EXAMPLE_DATA = pooch.create(
path=pooch.os_cache("scverse_tutorials"),
base_url="doi:10.6084/m9.figshare.22716739.v1/",
)
EXAMPLE_DATA.load_registry_from_doi()
pooch.create() : pooch 객체를 생성하여 데이터를 다운로드하는 역할
path = pooch.os_cache('scverse_tutorials') : 운영 체제의 캐시 디렉터리 내에 "scverse_tutorials"라는 폴더를 자동으로 생성
base_url = 'doi~' : 다운로드할 파일이 위치한 URL
load_registry_from_doi() : base_url에 지정된 DOI에서 파일 레지스트리(파일 목록)를 가져온다. 이 파일안에 pooch가 다운로드 해야 하는 파일 이름과 파일 해시 정보가 포함되어 있다.
samples = {
"s1d1": "s1d1_filtered_feature_bc_matrix.h5",
"s1d3": "s1d3_filtered_feature_bc_matrix.h5",
}
adatas = {}
for sample_id, filename in samples.items():
path = EXAMPLE_DATA.fetch(filename)
sample_adata = sc.read_10x_h5(path)
sample_adata.var_names_make_unique()
adatas[sample_id] = sample_adata
# EXAMPLE_DATA 는 pooch 객체
# EXAMPLE_DATA 는 로컬에서 캐시를 찾아서 경로설정
adata = ad.concat(adatas, label="sample")
adata.obs_names_make_unique()
print(adata.obs["sample"].value_counts())
adata
h5 확장자
- 보통 .h5 형식의 파일에는 유전자 ID, 세포 바코드, 발현 값 등이 포함됩니다.
- 파이썬의 Scanpy 라이브러리에서는 .h5 파일을 sc.read_10x_h5() 함수로 읽는다.
- 불러온 데이터는 anndata 객체로 저장한다.
path = EXAMPLE_DATA.fetch(filename) : 로컬에서 filename 경로를 찾아서 저장한다.
sample_adata = sc.read_10x_h5(path) : h5 데이터를 anndata 저장한다.
sample_adata.var_names_make_unique() : 유전자 이름 중복을 방지한다. ex) GAPDH-1, GAPDH-2
adatas[sample_id] = sample_adata : 각 색플의 AnnData 객체를 adatas 딕셔너리에 저장하는 역할
adata.obs_names_make_unique() : 중복된 세포 이름에 고유한 숫자를 추가하여 중복 제거
adata = ad.concat(adatas, label="sample") : 여러 개의 AnnData 객체를 하나의 통합된 객체로 결합, 즉 adatas에 여러개의 AnnData 객체를 결합함. label 은 각 샘플의 출처를 구분하는 레이블
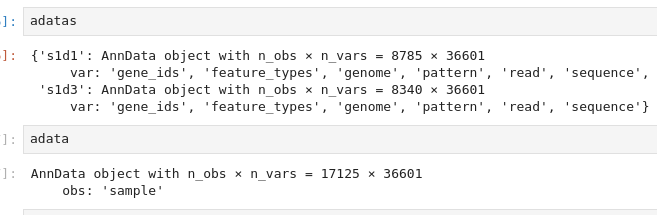
샘플 s1d1 8785개 , s1d3 8340개 합쳐서 17125개의 AnnData 객체 생성
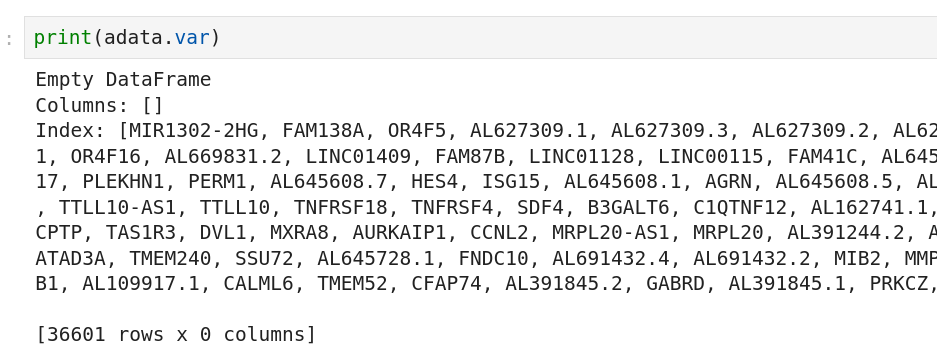
adata 출력하면 obs만 나오지만 adata.var로 var데이터에 접근할 수 있다.
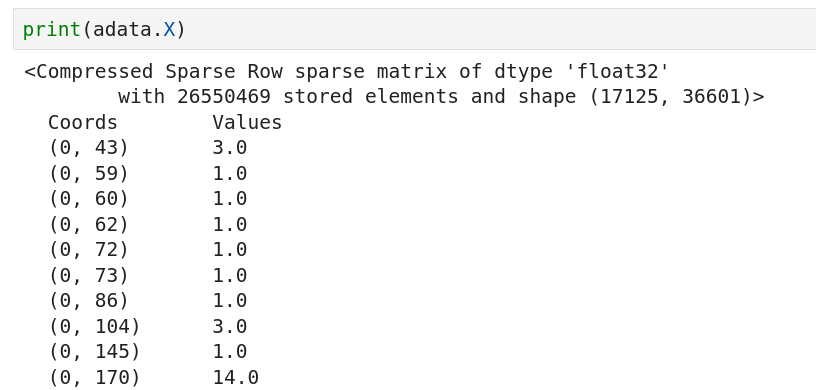
X 행렬의 구조
X행렬의 행은 obs의 인덱스를 따라 세포 순서가 결정되고, 열은 var의 인덱스를 따라 유전자 순서가 결정됨. X 행렬을 통해 세포별 유전자 발현 데이터를 obs와 var 메타데이터와 함께 저장함.
adata.obs['group'] = adata.obs['sample'].map({"s1d1": "control", "s1d3": "exp"})
s1d1을 control, s1d3를 exp 로 추가 ( obs는 cell 정보를 포함하는 메타데이터)
'bioinformatics' 카테고리의 다른 글
| Scanpy tutorials - preprocessing and clustering ( Doublet detection,Nomalization ) tutorial 코드 설명 (1) | 2024.11.12 |
|---|---|
| Scanpy tutorials - preprocessing and clustering ( Quality Control 개념설명) (4) | 2024.11.08 |
| 생명정보개론(bioinformatics) (1) | 2024.09.02 |
| 생명정보개론(bioinformatics) - Genebank,SNP 유전자 검색 (0) | 2024.08.14 |
| 생명정보개론(bioinformatics) - SNP, dbSNP (1) | 2024.08.13 |


