
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | ||
| 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| 13 | 14 | 15 | 16 | 17 | 18 | 19 |
| 20 | 21 | 22 | 23 | 24 | 25 | 26 |
| 27 | 28 | 29 | 30 |
- scanpy
- 오블완
- np.diagflat
- single cell rna sequening
- journal meeting
- pcrnaseq
- 티스토리챌린지
- 대학생재테크
- 대학생주식
- single cell rna sequencing
- 후기
- np.trace
- liver
- Tutorial
- singe cell ran sequencing
- 대학원
- Python
- R
- Preprocessing
- 통계학
- bioinformatics
- np.flatten
- endothelial
- scRNASeq
- np.triu
- 넘파이 매서드
- 선형대수
- 주식투자
- numpy
- 주식
- Today
- Total
biotechknowledge
Scanpy tutorials - preprocessing and clustering ( Doublet detection,Nomalization ) tutorial 코드 설명 본문
Scanpy tutorials - preprocessing and clustering ( Doublet detection,Nomalization ) tutorial 코드 설명
준2준2 2024. 11. 12. 22:12
Doublet detection
sc.pp.scrublet(adata, batch_key="sample")
Single cell rna sequencing은 세포 하나하나의 RNA 전사체를 분석한다. 세포 하나하나를 떼어내는 것은 기술적으로 굉장히 어렵기 때문에 세포 2개가 1개로 인식되는 경우가 있다. Scanpy는 doublet(세포2개) cell을 구별해내는 매서드 sc.pp.scrublet() 를 제공한다. (reference : graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells) scanpy 개발자들이 제공하는 툴들은 다 reference 논문의 내용을 코드로 옮긴 것이다.
위 코드를 실행하면 obs 에 'predicted_doublet' 컬럼이 만들어지고, 미리 짜여진 알고리즘에 따라 False, True(세포2개)를 구별 해낸다.
튜토리얼의 샘플 2개이다. s1d1, s1d3 각각 threshold가 다른 걸 확인할 수 있다. sc.pp.scrublet 함수에서는 doublet score의 분포를 바탕으로 최적의 threshold를 자동으로 결정한다.
import matplotlib.pyplot as plt
# 배치별 doublet score와 threshold 값을 가져와 시각화
batch_keys = adata.uns['scrublet']['batches'].keys() # 각 배치 이름 가져오기
plt.figure(figsize=(12, 6))
for i, batch in enumerate(batch_keys, start=1):
# 각 배치별 doublet score와 threshold 값 추출
doublet_scores = adata.obs.loc[adata.obs['sample'] == batch, 'doublet_score']
threshold = adata.uns['scrublet']['batches'][batch]['threshold']
# 히스토그램과 threshold 라인 그리기
plt.subplot(1, len(batch_keys), i)
plt.hist(doublet_scores, bins=50, density=True, alpha=0.75, color='purple', label="Doublet Scores")
plt.axvline(threshold, color='red', linestyle='dashed', linewidth=2, label=f"Threshold: {threshold:.3f}")
plt.title(f"Batch {batch} - Doublet Scores")
plt.xlabel("Doublet Score")
plt.ylabel("Density")
plt.legend()
plt.tight_layout()
plt.show()각각의 배치별로 Double Scores 히스토그램을 그리고 각각의 Threshold를 표시하였다.
Normalization
Cell Depth Scaling : 각 세포의 측정된 유전자 별현값이 세포마다 다르다. 유전자 발현 수준이 다르기 때문에, 이를 비교 가능하게 만들기 위해 데이터를 정규화 해야 한다.
# Saving count data
adata.layers["counts"] = adata.X.copy()
원본 카운트 데이터 ( scRNA-seq (single-cell RNA sequencing) 실험에서 각 세포에서 측정된 유전자 발현량을 나타내는 처리되지 않은 데이터) 를 나타낸다. 유전자 발현 카운트 데이터('adata.X') 를 복사하여 'adata.layers['counts'] 에 저장하는 역할은 한다. 나중에 다시 불러 올 때 adata.X = adata.layers['counts'].copy() 로 원본 복구 가능.
# Normalizing to median total counts
sc.pp.normalize_total(adata)
# Logarithmize the data
sc.pp.log1p(adata)
세포 유전자 발현양을 정규화하는 코드이다. 세포별로 유전자 발현 총량을 맞추어 비교 가능하도록 만든다.
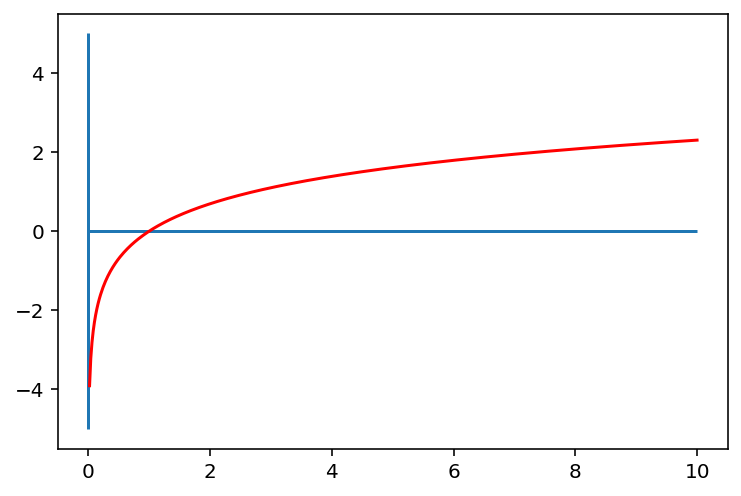
로그 변환 하는 이유 : 0에 가깝게 모여있는 값들이 x로 입력되면, 그 함수값인 y 값들은 매우 큰 범위로 벌어지게 된다.
즉, 로그함수는 0에 가까운 값들이 조밀하게 모여있는 입력값을 넓은 범위로 펼치는 의미를 갖는다. 데이터의 분포를 균일하게 만들어 분석에 적합한 형태로 만든다.
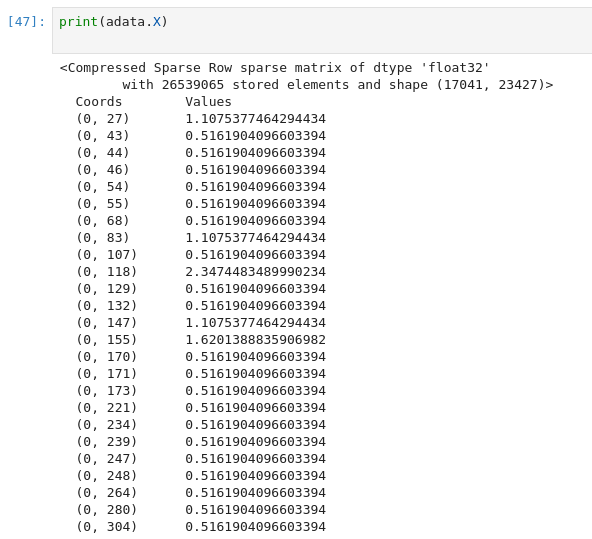
정규화 후 value 값이 log scale 로 변환된 것을 확인할 수 있다.
Feature selection
유전자 수가 굉장히 많기 때문에 데이터 분석에 도움이 되지 않는 유전자가 많다. 각 유전자의 변동성을 계산하여 중요한 유전자들을 찾아주어야 한다. pp.highly_variable_genes() 함수를 이용한다. 크게 3가지 방식이 있는데 매개변수로 지정해 줄 수 있다. ( Seurat, Cell Ranger, seurat v3)
sc.pp.highly_variable_genes(adata, n_top_genes=2000, batch_key="sample")
batch별로, 변동성이 큰 2000개의 유전자만 사용한다. 노이즈와 데이터차원을 줄이는 과정이다.
sc.pl.highly_variable_genes(adata)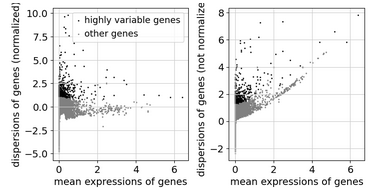
위그래프는 각각 정규화 분산과 비정규화분산에 따른 highly variable genes 를 나타낸 것이다. 통계적으로 평균이 높은 경우에 분산이 커지는 경향이 있다. 그렇게 때문에 hige variable을 찾는데 있어 방해가 되기 때문에 보정해줘야 한다. 분산을 보정하는 통계적인 여러 방식이 있는데 sc.pp.highly_variable_genes() 함수를 이용하면 딸깍으로 해결할 수 있다.



