
| 일 | 월 | 화 | 수 | 목 | 금 | 토 |
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||
| 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 12 | 13 | 14 | 15 | 16 | 17 | 18 |
| 19 | 20 | 21 | 22 | 23 | 24 | 25 |
| 26 | 27 | 28 | 29 | 30 | 31 |
- single cell rna sequencing
- 후기
- 대학생주식
- np.triu
- 대학생재테크
- endothelial
- np.diagflat
- 통계학
- 티스토리챌린지
- R
- np.trace
- np.flatten
- 대학원
- Python
- Preprocessing
- scanpy
- liver
- scRNASeq
- single cell rna sequening
- bioinformatics
- 연구원
- 오블완
- 주식투자
- Tutorial
- 주식
- 넘파이 매서드
- numpy
- 선형대수
- journal meeting
- pcrnaseq
- Today
- Total
biotechknowledge
Scanpy tutorials - data integration, injest 코드 개념 설명 본문

생물학 논문의 기본은 실험군과 대조군을 비교하는 것이다. 예를들면 카페인을 많이 마시면 머리가 나빠진다고 주장하고싶으면 커피를 매일 마신 그룹과 물을 매일 마신 그룹을 비교하면 된다. scRNAseq도 마찬가지이다. scRNAseq은 대조군 만으로도 도출해낼 수 있는 정보가 많지만, 실험군과 비교하였을때 얻어낼 수 있는 정보의 양과 질을 극대화할 수 있다. Integration 이란, 비교하고 싶은 두 그룹을 하나의 벡터공간상에 투여하는 것을 말한다. 두 그룹은 같은 벡터공간에 존재하게되어 cell cluster를 직접 비교할 수 있고 그룹별 batch effect 보정을 하는 알고리즘,통계 기법의 발전으로 신뢰도를 더해가고 있다.
import scanpy as sc
import pandas as pd
sc.settings.verbosity = 2 # verbosity: errors (0), warnings (1), info (2), hints (3)
sc.logging.print_versions()
sc.settings.set_figure_params(dpi=80, frameon=False, figsize=(3, 3), facecolor="white")
코드의 진행정보를 얻기 위해 verbosity 2 로설정한다. set_figure_params 는 그래프 모양설정이다.
PBMCs
# this is an earlier version of the dataset from the pbmc3k tutorial
adata_ref = sc.datasets.pbmc3k_processed()
adata = sc.datasets.pbmc68k_reduced()
scanpy에서 제공하는 연습용 데이터를 불러온다. adata_ref 와 adata는 integration에 사용할 두 데이터셋이다. 보통 control을 adata_ref에 실험군을 adata에 불러온다. adata_ref 데이터는 분석을 완료한 control 이고 metadata, PCA, clustering 결과가 포함되어 있다.
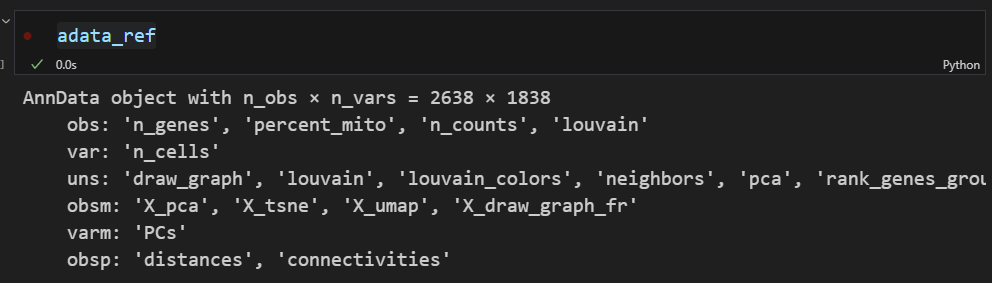
louvain 알고리즘으로 클러스터링되어있다. 최근에는 leiden을 더 많이 사용한다. adata_ref.obs['louvain'] 에는 cell type annotation이 저장되어 있다. adata를 adata_ref 벡터공간에 투영하고 clustering pocess를 따라간다.
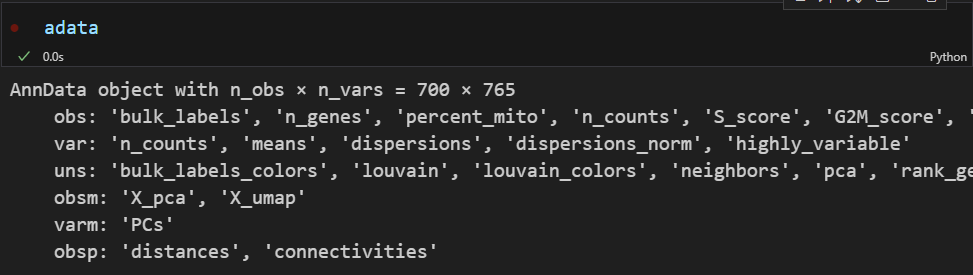
adata 에는 700개의 cell이 있다. 위 데이터는 연습용데이터로 pbmc68k에서 일부 가져온 것으로 실전이었으면 obsm 과 몇 obs컬럼은 텅 비어있는게 정상이다.
var_names = adata_ref.var_names.intersection(adata.var_names)
adata_ref = adata_ref[:, var_names]
adata = adata[:, var_names]
두 데이터를 합쳐야하게때문에 두 데이터셋에서 공통으로 발현된 유전자만 걸러온다.
sc.pp.pca(adata_ref)
sc.pp.neighbors(adata_ref)
sc.tl.umap(adata_ref)
adata_ref 로 PCA 진행한다.
sc.pl.umap(adata_ref, color="louvain")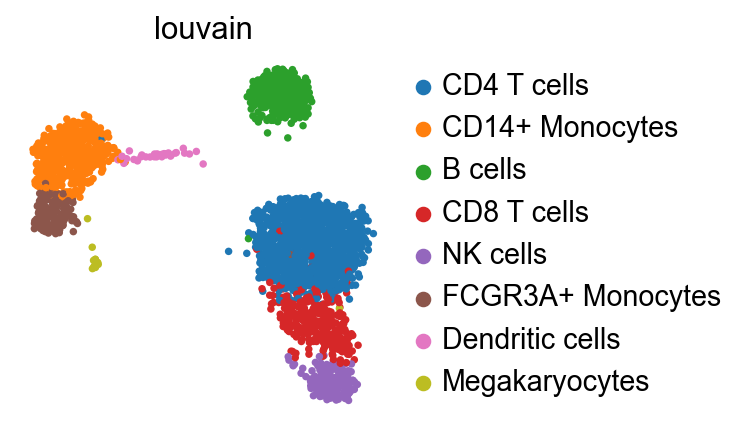
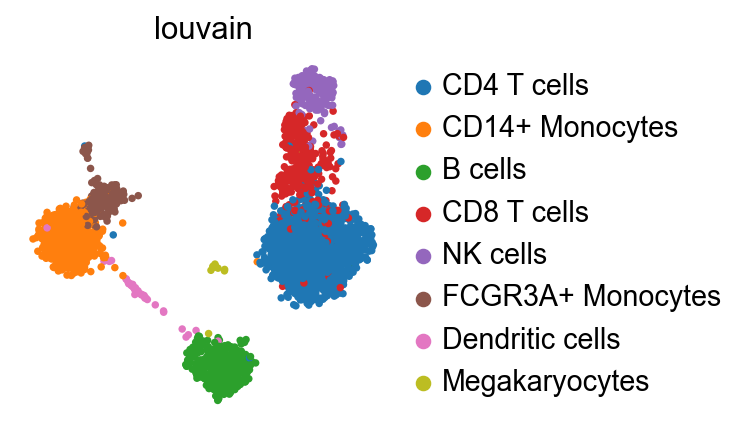
왼쪽이 원래 ref가 가지고있던 정보로 UMAP을 그린 것이고 오른쪽은 공통 유전자만 남기고 UMAP을 그린 것이다. 유전자가 1838개에서 208개로 줄었음에도 clustering 결과를 잘 유지하고 있는걸 확인 할 수 있다. 유전체 데이터는 sparsity가 심하기 때문에 어차피 많은 수의 유전자가 허수이다. 그래서 제거해도 결과가 비슷한걸로 추측해볼 수 있다.
Mapping PBMCs using ingest
sc.tl.ingest(adata, adata_ref, obs="louvain")
adata_ref 의 클러스터 라벨(louvain)과 임베딩 정보를 adata에 매핑한다. adata의 클러스터 라벨을 예측하고 동일한 UMAP 좌표계를 공유한다.
adata.uns["louvain_colors"] = adata_ref.uns["louvain_colors"] # fix colors
클러스터 라벨의 색상을 adata_ref 와 동일하게 설정한다.
sc.pl.umap(adata, color=["louvain", "bulk_labels"], wspace=0.5)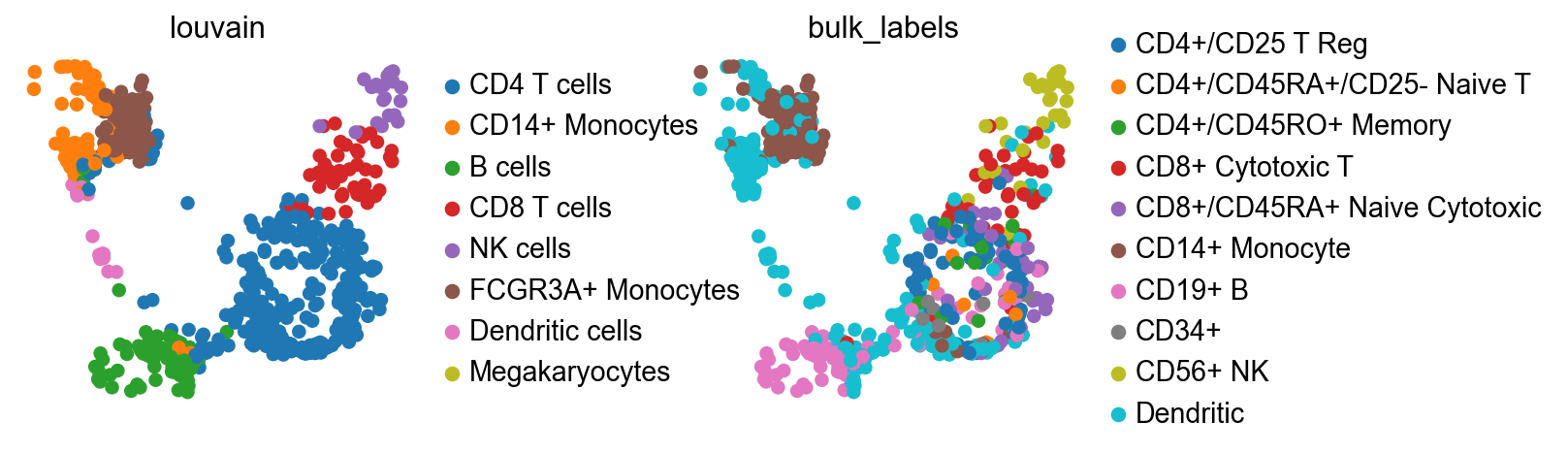
injest 를 통해 annotation 한 결과 왼쪽과 원래 갖고있던 정보(연습용 dataset으로 세포 타입이 bulk_labels에 저장되어 있음)를 비교해 볼 수 있다. 왼쪽 위를 보면 갈색은 비슷한 위치에 매핑되었고 주황색과 하늘색은 Myeroid lineage로 넓은 의미에서 비슷한 계열로 정확하게 짚어내진 못한 걸 확인할 수 있다.
adata_concat = adata_ref.concatenate(adata, batch_categories=["ref", "new"])
adata 와 ref 를 하나의 객체로 합치고 batch 카탈로그를 부여한다.
adata_concat.obs.louvain = adata_concat.obs.louvain.astype("category")
# fix category ordering
adata_concat.obs.louvain.cat.reorder_categories(
adata_ref.obs.louvain.cat.categories, inplace=True
)
# fix category colors
adata_concat.uns["louvain_colors"] = adata_ref.uns["louvain_colors"]
concat 객체에서 louvain을 caterogy 로 바꿔준다. ref 에 저장된 카테고리 순서를 따라 저장한다. 클러스터의 색상 정보를 ref에서 복사하여 클러스터 색상이 일관되게 유지되도록 한다.
sc.pl.umap(adata_concat, color=["batch", "louvain"])

Monocyte에서 batch effect 가 있다.(cluster가 대부분 ref로 이우어져 있다.) Megakaryocyte는 ref에만 존재하는 cluster로 데이터 크기가 작아서 생겨서 생긴 일이다. 만약 ref 와 adata가 뒤바뀌었다면 이 세포타입은 사라진다.
다음편에서 Normalzation, scaling, BBKNN 계속..



